About
Background
In general, co-expressed genes tend to have associated transcriptional regulatory mechanisms and involve in similar metabolic pathways. Since transcription factor is a critical regulator of gene expression, it is worth to investigate TFs on the promoter of co-expressed genes. Although co-expressed genes and their related metabolic pathways could be easily identified from previous resources such as EXPath and EXPath Tool, none of them are available to obtain their regulating TFs simultaneously. Moreover, organ/tissue/stage- specific genes cannot be retrieved from those platforms. |
Description
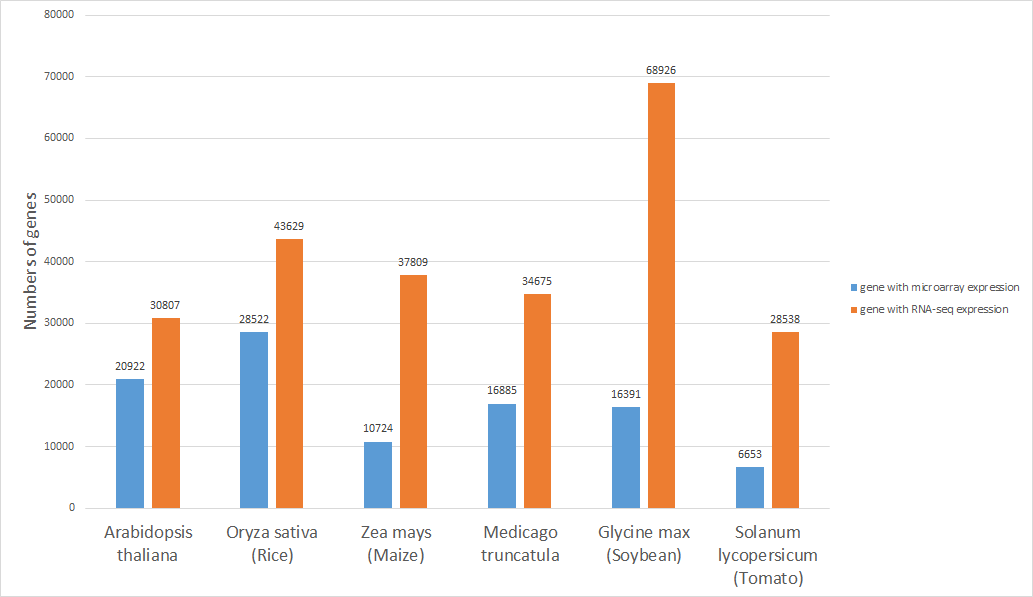
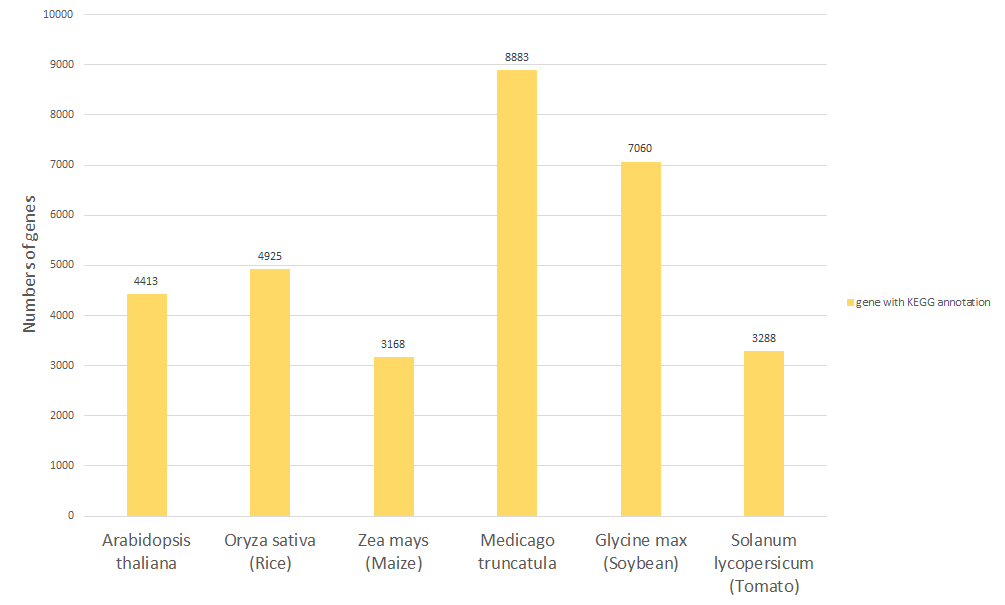
EXPath 2.0 serves as an updated database for the realization of regulatory mechanisms in various plant metabolic pathways. In the current version, 1881 microarray samples from various developmental stages and stresses from six plants including Arabidopsis, rice, maize, Medicago, soybean and tomato were integrated, respectively. There are four significant developments different from previous version. (i) high-throughput data from three crops, including medicago, soybean and tomato is added. (ii) stage-specific genes from various developmental stages are available. (iii) a correlation network according to a group of input genes from users can be constructed. (iv) TFs of a group of genes in a metabolic pathway or co-expression network can be retrieved. (v) Comparative analysis of metabolic pathways and TF regulatory mechanisms in an input gene group are available. |
Conclusions
EXPath 2.0 is a conveniently resource for investigating gene expression in metabolic pathways under specific conditions and facilitates users to access the regulatory mechanisms of the critical bio-pathways from plant high-throughput data. This database is freely available at http://EXPath.itps.ncku.edu.tw. |
The number of microarray samples integrated in Expath
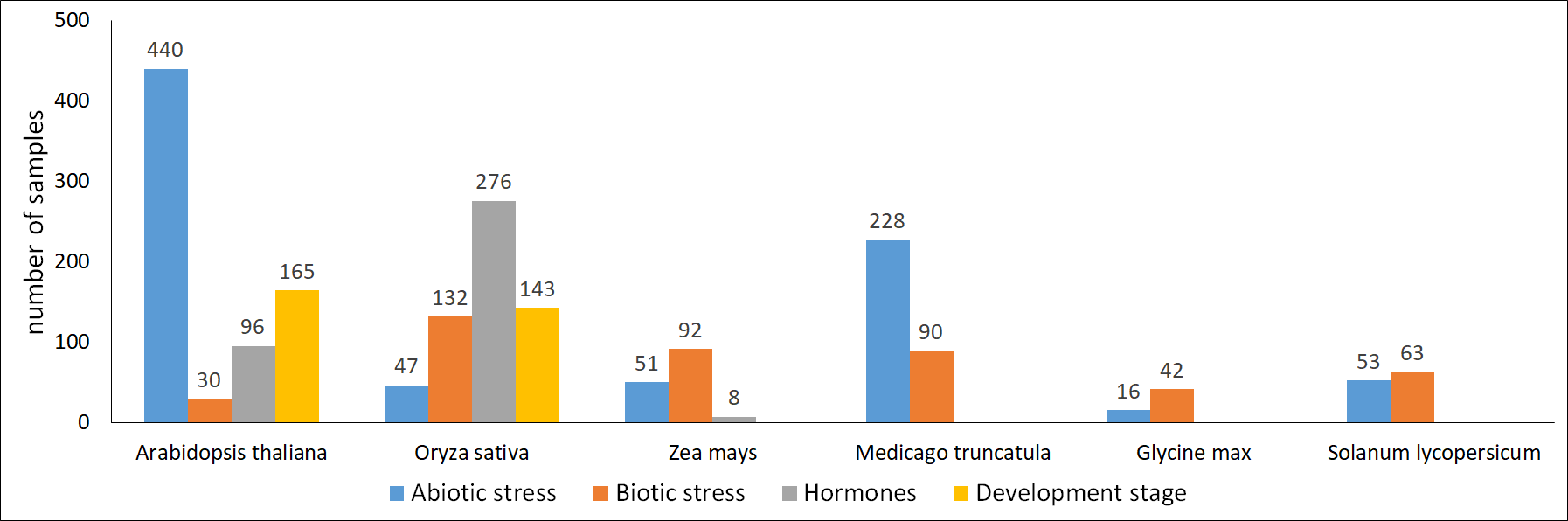 |
The number of RNA-seq samples integrated in Expath
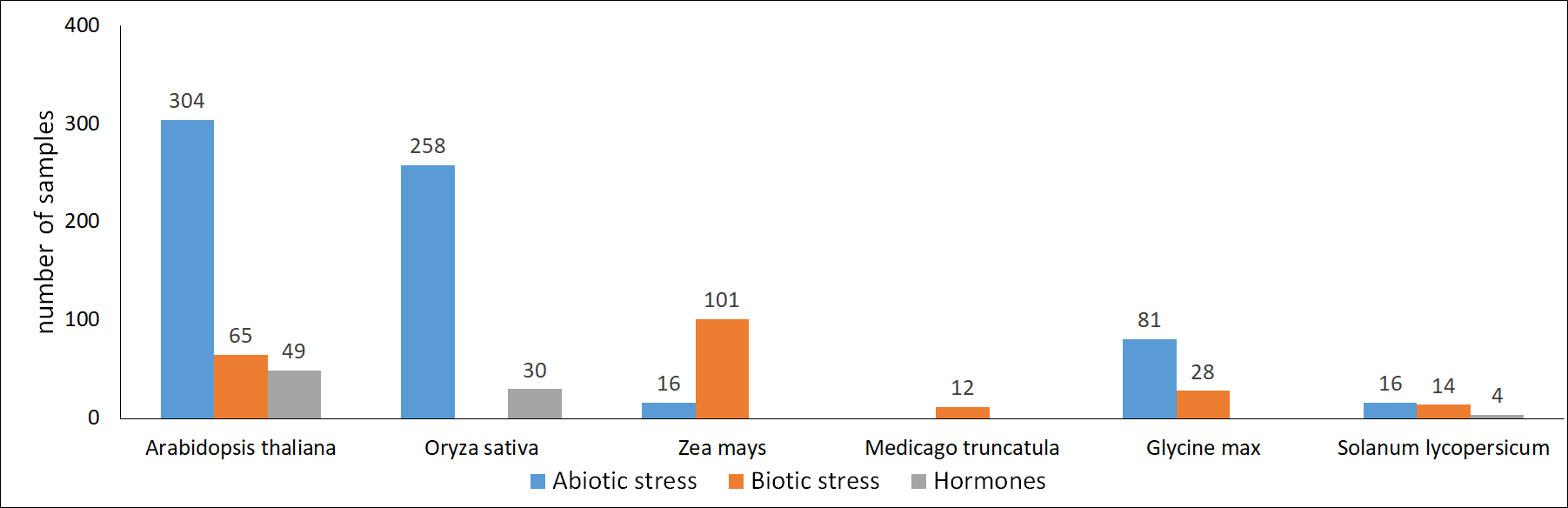 |
Categories of microarray samples in EXPath expression database
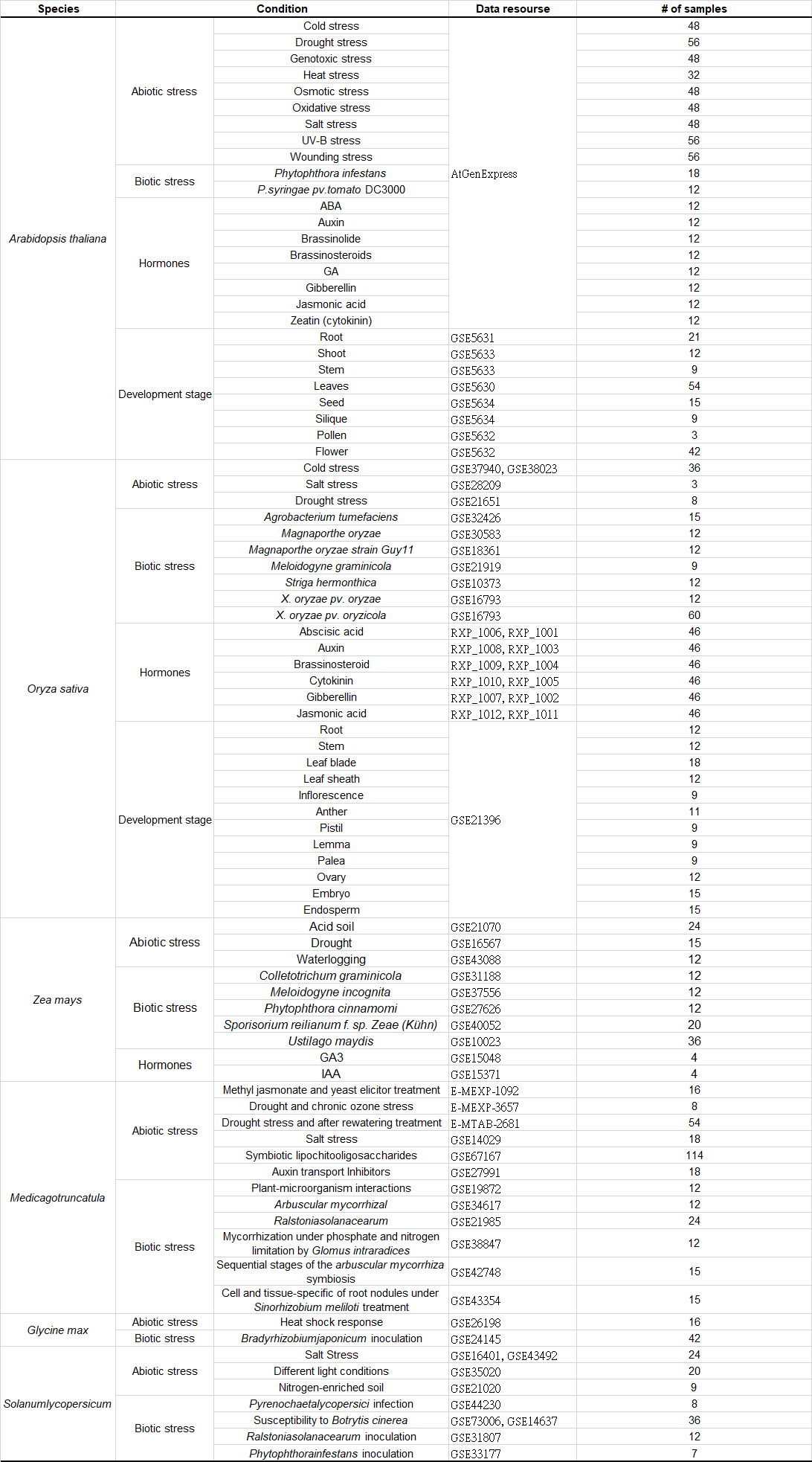 |
Categories of RNA-seq samples in EXPath expression database
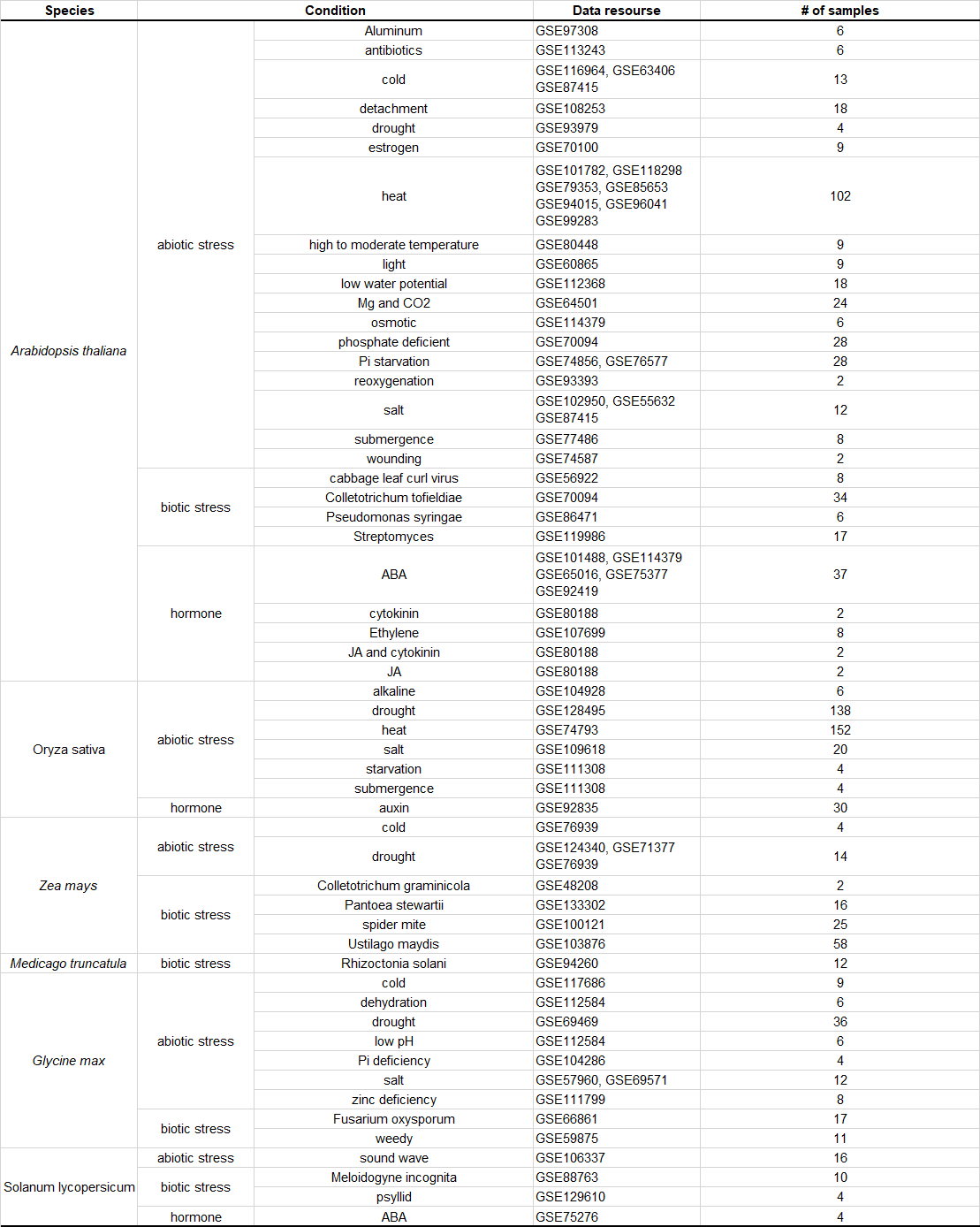 |
The number of genes collected in Expath
  |
